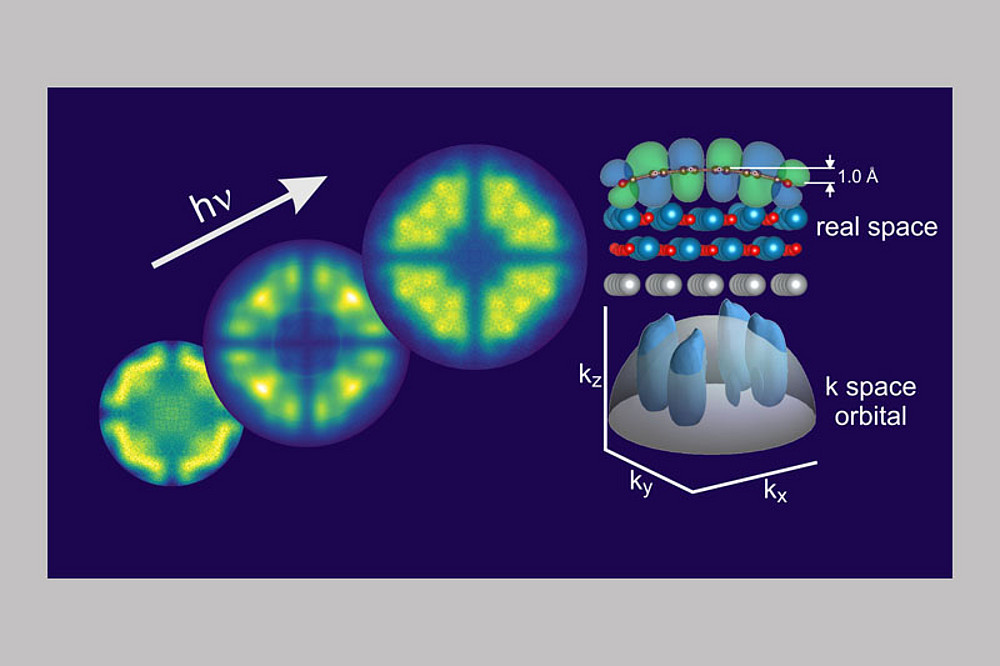
Die geometrische Form von kondensierten aromatischen Molekülen wird durch sp2-Hybridisierung bestimmt, was zu einem planaren Netzwerk von Bindungen führt, deren elektronische Struktur durch eine Reihe von σ-Orbitalen und π-Orbitalen gekennzeichnet ist. Letztere zeichnen sich durch Elektronendichte auf beiden Seiten der Molekülebene aus. Aufgrund dieser elektronischen Konfiguration besitzt das Kohlenstoffgerüst solcher Moleküle eine gewisse Steifigkeit. Beispiele hierfür sind etwa kleine Moleküle wie Benzol, aber auch größere ein- und zweidimensionale Strukturen wie die linearen Acene oder Coronen, bis hin zu Graphen. Obwohl diese Moleküle vergleichsweise resistent gegen geometrische Verzerrungen sind, sind letztere oft sogar wünschenswert, da sie Auswirkungen auf die chemischen und optoelektronischen Eigenschaften haben können.
In einer vor Kurzem in der Fachzeitschrift ACS Nano veröffentlichten Publikation, resultierend aus einer Kooperation der Surface Science und Computational Physics-Gruppen der Uni Graz mit Partnern von FZ Jülich und PTB Berlin, wurde gezeigt, dass das molekulare Gerüst des planaren Moleküls PTCDA (Perylenetetracarbonsäuredianhydrid) bei der Adsorption auf einer dünnen Magnesiumoxid (MgO)-Schicht stark gekrümmt wird. Es stellte sich die Frage, wie diese Krümmung quantitativ bestimmt werden kann. Dazu nutzten die Forscher die Photoemissions-Orbital-Tomographie (POT)-Methode, die Bilder der elektronischen Molekülzustände im Impulsraum liefert. Die Experimente dazu wurden zum Teil an Synchrotron-Strahlungsquellen durchgeführt, zum Teil auch an der NAWI Graz core facility NanoPEEM. Im Speziellen machten sich die Forscher die Photonenenergie-Abhängigkeit der Impulsraumverteilung der aus den elektronischen Zuständen emittierten Photoelektronen zunutze, um im Vergleich mit Simulationen basierend auf der Dichtefunktionaltheorie (DFT) quantitative Aussagen über die Krümmung des Moleküls zu machen. Bemerkenswert dabei ist, dass die bestimmte Krümmung des Moleküls auf dem dielektrischen MgO-Substrat viel größer ist als auf einer Silber-Oberfläche, die zu Vergleichszwecken untersucht wurde. Dieser Umstand lässt sich durch die schwächere Polarisierbarkeit und damit größere elektronische Härte des MgO erklären. Diese Arbeit zeigt die Vorteile der POT-Methode gegenüber anderen Methoden bei der Bestimmung von geometrischen Eigenschaften von adsorbierten Molekülen auf.
Zitat:
Philipp Hurdax, Christian S. Kern, Thomas Georg Boné, Anja Haags, Michael Hollerer, Larissa Egger, Xiaosheng Yang, Hans Kirschner, Alexander Gottwald, Mathias Richter, François C. Bocquet, Serguei Soubatch, Georg Koller, Frank Stefan Tautz, Martin Sterrer, Peter Puschnig,and Michael G. Ramsey
Large Distortion of Fused Aromatics on Dielectric Interlayers Quantified by Photoemission Orbital Tomography.
ACS Nano 2022, 16, 17435–17443. https://doi.org/10.1021/acsnano.2c08631